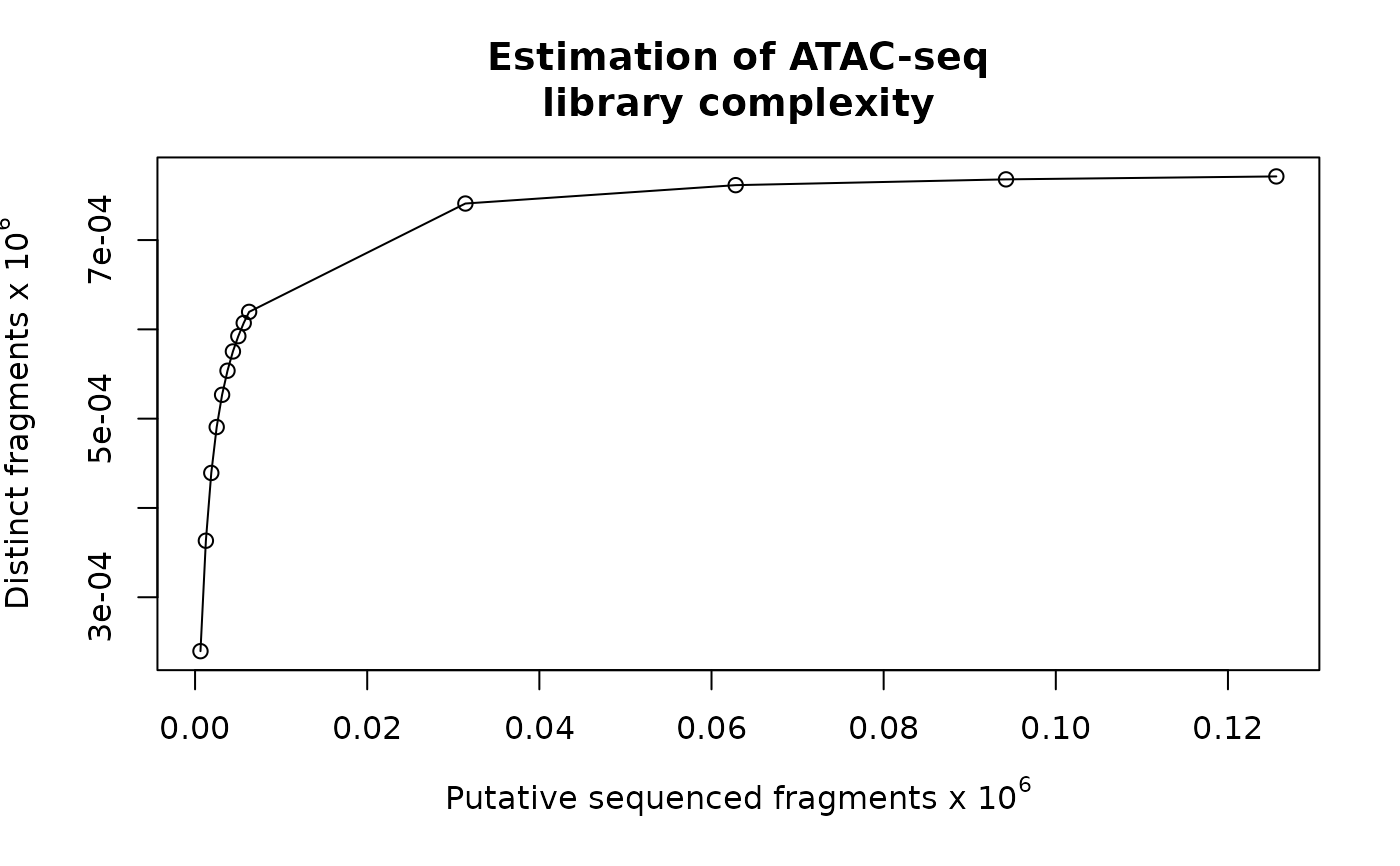
Library complexity estimation
estimateLibComplexity.RdEstimating the library complexity.
estimateLibComplexity( histFile, times = 100, interpolate.sample.sizes = seq(0.1, 1, by = 0.1), extrapolate.sample.sizes = seq(5, 20, by = 5) )
Arguments
| histFile | A two-column matrix of integers. The 1st column is the frequency j = 1,2,3,.... The 2nd column is the number of genomic regions with the same fequency (j) of duplication. This file should be sorted by the first column in ascending order. For example, one row of a histogram file: 10 20 means there are 10 genomic regions, each of which is covered by 20 identical fragments at a given sequencing depth of a sequencing library. |
|---|---|
| times | An positive integer representing the minimum required number of successful estimation. Default is 100. |
| interpolate.sample.sizes | A numeric vector with values between (0, 1]. |
| extrapolate.sample.sizes | A numeric vector with values greater than 1. |
Value
invisible estimates, a data frame of 3 columns: relative sequence depth, number of distinct fragments, number of putative sequenced reads.
See also
Author
Haibo Liu, Feng Yan