Introduction
The HMMtBroadPeak package is designed to call very broad peaks for data such as lamina-associated domains (LADs), nucleolus-associated domains (NADs), or other topologically associating domains.
The methods is following the description of Christ et.al1. Reads will be count by each bins. Only bins with at least given reads (defined by background parameter) for all samples (pool all reads for each bin) will be subsequently normalized. These bins will be first normalized to CPM (count per million) reads and then do log2 transform for the ratio over control with a pseudocount. The peaks were defined by running a hidden markov model over the normalized values (using the R-package HMMt).
Quick start
There are three steps for calling peaks:
Step1: prepare the bam files.
The bam files should be clean with reads passed quality control and proper paired (if applicable). The index file of bam should be stored in the same folder and with same prefix.
treatment <- system.file("extdata", "LB1.KD.chr1_1_5000000.bam",
package = "HMMtBroadPeak",
mustWork = TRUE)
control <- system.file("extdata", "LB1.WT.chr1_1_5000000.bam",
package = "HMMtBroadPeak",
mustWork = TRUE)
## For local file, please try
# treatment <- "path/to/treatment/bam/files"
# control <- "path/to/control/bam/files"Step2: calling peaks.
The reads counts for treatment and control will be pool for each group. That is to say duplicates will not be considered when we call peaks.
library(HMMtBroadPeak)
called <- HMMtBroadPeak(treatment, control)## iteration: 1iteration: 2iteration: 3iteration: 4iteration: 5iteration: 6iteration: 7iteration: 8iteration: 9iteration: 10iteration: 11iteration: 12iteration: 13iteration: 14iteration: 15iteration: 16iteration: 17iteration: 18iteration: 19iteration: 20iteration: 21iteration: 22iteration: 23iteration: 24iteration: 25iteration: 26iteration: 27iteration: 28iteration: 29iteration: 30iteration: 31iteration: 32iteration: 33iteration: 34iteration: 35iteration: 36iteration: 37iteration: 38iteration: 39iteration: 40iteration: 41iteration: 42iteration: 43iteration: 44iteration: 45iteration: 46iteration: 47iteration: 48iteration: 49iteration: 50iteration: 51iteration: 52iteration: 53iteration: 54iteration: 55
called$peaks## GRanges object with 9 ranges and 0 metadata columns:
## seqnames ranges strand
## <Rle> <IRanges> <Rle>
## [1] chr1 499502-504496 *
## [2] chr1 594407-609391 *
## [3] chr1 624377-649351 *
## [4] chr1 664337-669331 *
## [5] chr1 709292-719281 *
## [6] chr1 729272-734266 *
## [7] chr1 754247-2662339 *
## [8] chr1 2672330-2692309 *
## [9] chr1 2747255-5000001 *
## -------
## seqinfo: 1 sequence from an unspecified genome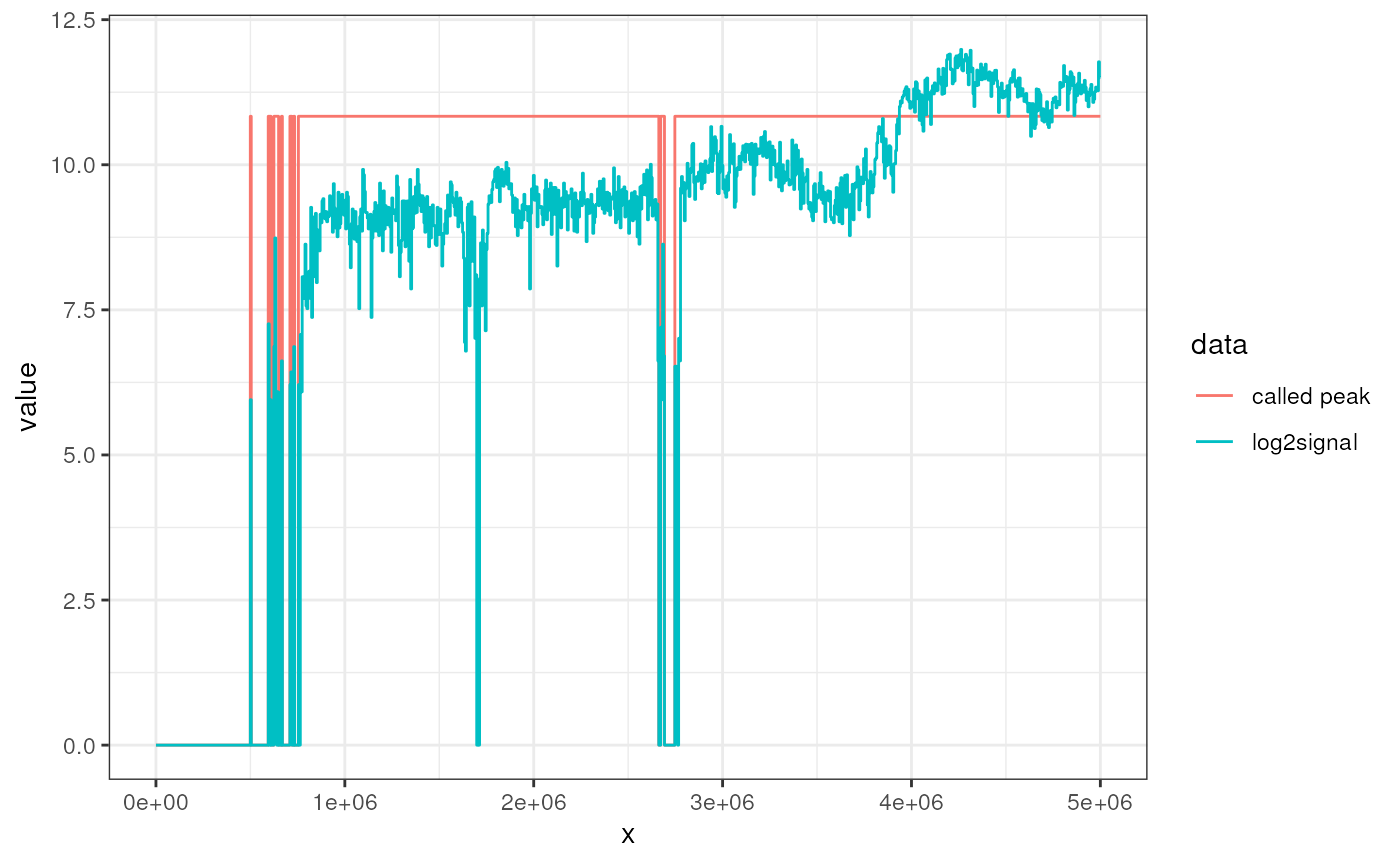
Session Info
## R version 4.4.1 (2024-06-14)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 22.04.4 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.20.so; LAPACK version 3.10.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## time zone: Etc/UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] ggplot2_3.5.1 rtracklayer_1.65.0
## [3] HMMtBroadPeak_0.0.5 GenomicAlignments_1.41.0
## [5] Rsamtools_2.21.0 Biostrings_2.73.1
## [7] XVector_0.45.0 SummarizedExperiment_1.35.1
## [9] Biobase_2.65.0 MatrixGenerics_1.17.0
## [11] matrixStats_1.3.0 GenomicRanges_1.57.1
## [13] GenomeInfoDb_1.41.1 IRanges_2.39.1
## [15] S4Vectors_0.43.1 BiocGenerics_0.51.0
##
## loaded via a namespace (and not attached):
## [1] gtable_0.3.5 rjson_0.2.21 xfun_0.45
## [4] bslib_0.7.0 htmlwidgets_1.6.4 lattice_0.22-6
## [7] vctrs_0.6.5 tools_4.4.1 bitops_1.0-7
## [10] curl_5.2.1 parallel_4.4.1 tibble_3.2.1
## [13] fansi_1.0.6 highr_0.11 pkgconfig_2.0.3
## [16] Matrix_1.7-0 desc_1.4.3 lifecycle_1.0.4
## [19] GenomeInfoDbData_1.2.12 farver_2.1.2 compiler_4.4.1
## [22] textshaping_0.4.0 munsell_0.5.1 codetools_0.2-20
## [25] htmltools_0.5.8.1 sass_0.4.9 RCurl_1.98-1.16
## [28] yaml_2.3.9 pkgdown_2.1.0 pillar_1.9.0
## [31] crayon_1.5.3 jquerylib_0.1.4 BiocParallel_1.39.0
## [34] DelayedArray_0.31.7 cachem_1.1.0 abind_1.4-5
## [37] HMMt_0.1 digest_0.6.36 restfulr_0.0.15
## [40] labeling_0.4.3 fastmap_1.2.0 grid_4.4.1
## [43] colorspace_2.1-0 cli_3.6.3 SparseArray_1.5.20
## [46] magrittr_2.0.3 S4Arrays_1.5.4 XML_3.99-0.17
## [49] utf8_1.2.4 withr_3.0.0 UCSC.utils_1.1.0
## [52] scales_1.3.0 rmarkdown_2.27 httr_1.4.7
## [55] ragg_1.3.2 evaluate_0.24.0 knitr_1.48
## [58] BiocIO_1.15.0 rlang_1.1.4 glue_1.7.0
## [61] jsonlite_1.8.8 R6_2.5.1 systemfonts_1.1.0
## [64] fs_1.6.4 zlibbioc_1.51.1