trackViewer Vignette: change the track styles
Jianhong Ou, Lihua Julie Zhu
5 September 2024
changeTracksStyles.RmdAbstract
Visualize mapped reads along with annotation as track layers for NGS dataset such as ChIP-seq, RNA-seq, miRNA-seq, DNA-seq, SNPs and methylation data.
Plot high dense methylation/mutation/variant data by dandelion plot
Plot genomic interaction data (HiC, cooler, etc) by viewTracks
Prepare toy data
First, we import the sample data.
library(trackViewer)
extdata <- system.file("extdata", package="trackViewer",
mustWork=TRUE)
repA <- importScore(file.path(extdata, "cpsf160.repA_-.wig"),
file.path(extdata, "cpsf160.repA_+.wig"),
format="WIG")
## Because the wig file does not contain any strand info,
## we need to set it manually.
strand(repA$dat) <- "-"
strand(repA$dat2) <- "+"
fox2 <- importScore(file.path(extdata, "fox2.bed"), format="BED",
ranges=GRanges("chr11", IRanges(122830799, 123116707)))
dat <- coverageGR(fox2$dat)
## We can split the data by strand into two different track channels
## Here, we set the dat2 slot to save the negative strand info.
fox2$dat <- dat[strand(dat)=="+"]
fox2$dat2 <- dat[strand(dat)=="-"]
library(TxDb.Hsapiens.UCSC.hg19.knownGene)
library(org.Hs.eg.db)
gr <- GRanges("chr11", IRanges(122929275, 122930122), strand="-")
trs <- geneModelFromTxdb(TxDb.Hsapiens.UCSC.hg19.knownGene,
org.Hs.eg.db,
gr=gr)
optSty <- optimizeStyle(trackList(repA, fox2, trs))
trackList <- optSty$tracks
viewerStyle <- optSty$styleUsing the browseTracks function as a helper
Since version 1.33.6, the interactive plot generated by
browseTracks function will print the psuedocode about how
to change the track styles at the bottom, which can be used as a quick
reference.
browseTracks(trackList, gr=gr)Adjust the x-axis or the X scale
In most cases, researchers are interested in the relative position of the peaks in the gene. Sometimes, margin needs to be adjusted to be able to show the entire gene model. The Figure below shows how to add an X scale (x-scale) and remove the x-axis using the setTrackXscaleParam and setTrackViewerStyleParam functions.
setTrackViewerStyleParam(viewerStyle, "xaxis", FALSE)
setTrackViewerStyleParam(viewerStyle, "margin", c(.01, .05, .01, .01))
setTrackXscaleParam(trackList[[1]], "draw", TRUE)
setTrackXscaleParam(trackList[[1]], "gp", list(cex=0.8))
viewTracks(trackList, gr=gr, viewerStyle=viewerStyle)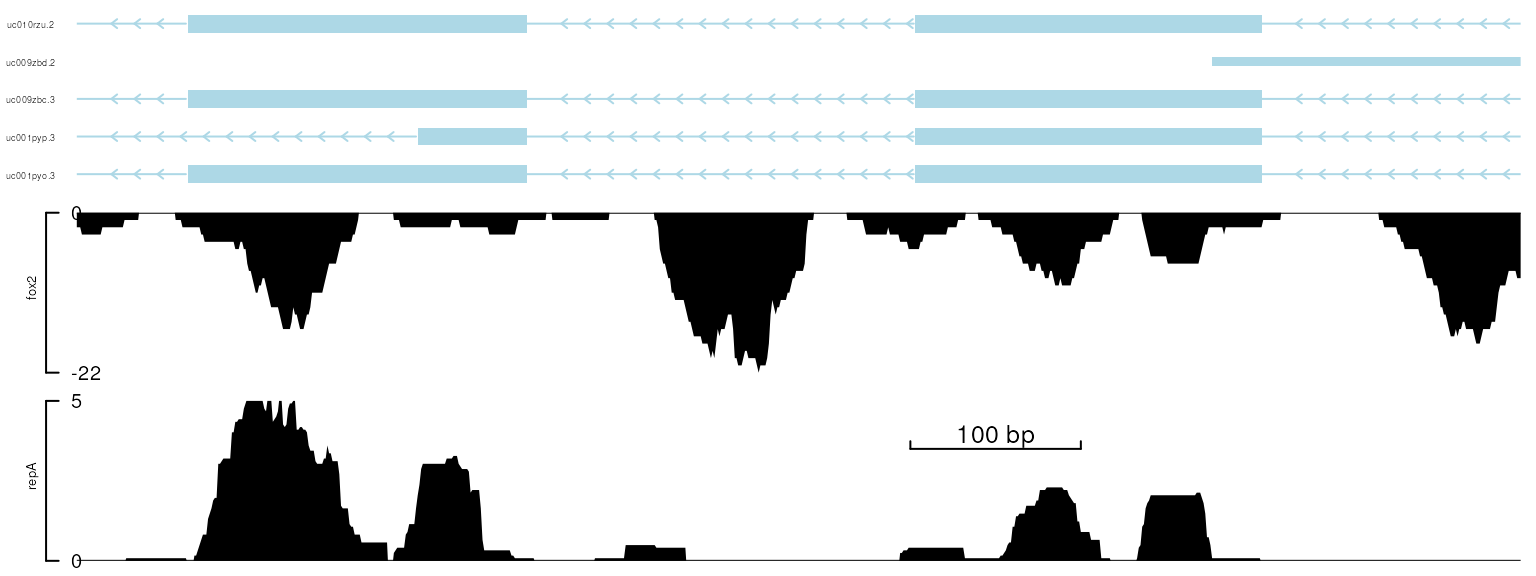
plot data with x-scale
setTrackXscaleParam(trackList[[1]], attr="position",
value=list(x=122929700, y=3, label=200))
viewTracks(trackList, gr=gr, viewerStyle=viewerStyle)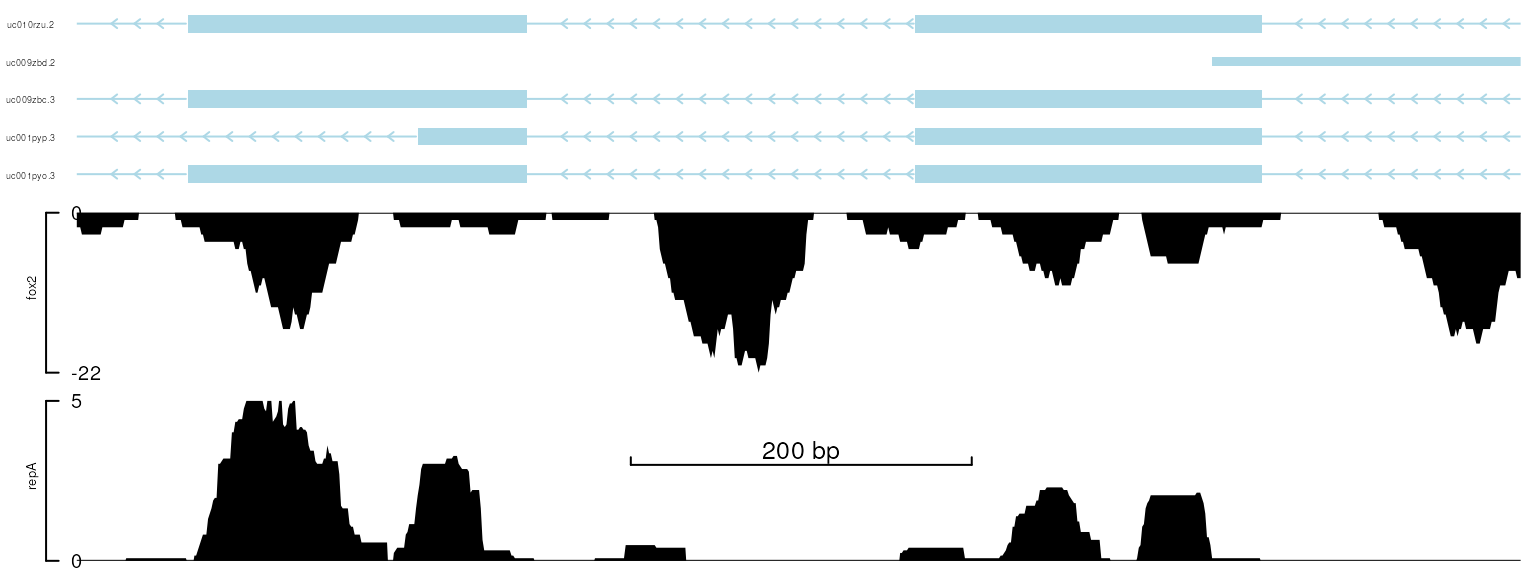
plot data with x-scale
Adjust the y-axis
The y-axis can be put to the right side of the track by setting the main slot to FALSE in the y-axis slot of each track. In addition, the limit of y-axis (ylim) can be set by setTrackStyleParam.
setTrackViewerStyleParam(viewerStyle, "margin", c(.01, .05, .01, .05))
for(i in 1:2){
setTrackYaxisParam(trackList[[i]], "main", FALSE)
}
## Adjust the limit of y-axis
setTrackStyleParam(trackList[[1]], "ylim", c(0, 25))
setTrackStyleParam(trackList[[2]], "ylim", c(-25, 0))
## The y-axis tick labels can be turn off/on by setting the 'label' to FALSE/TRUE
#setTrackYaxisParam(trackList[[1]], 'label', TRUE)
viewTracks(trackList, gr=gr, viewerStyle=viewerStyle)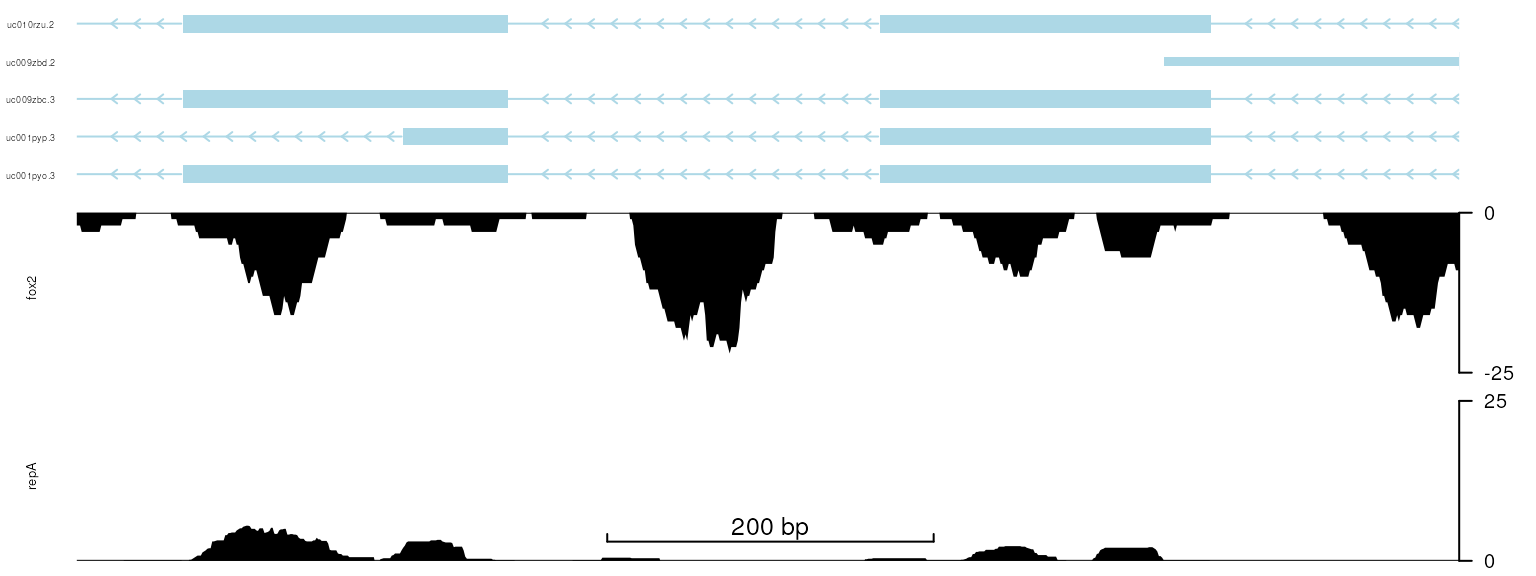
plot data with y-axis in right side
The function setTrackYaxisParam can be sued to adjust
the track color/size/etc of y-axis and the y-axis labels.
## change the
setTrackYaxisParam(trackList[[1]], "gp", list(cex=.8, col="green"))
setTrackYaxisParam(trackList[[2]], "gp", list(cex=.8, col="blue"))
viewTracks(trackList, gr=gr, viewerStyle=viewerStyle)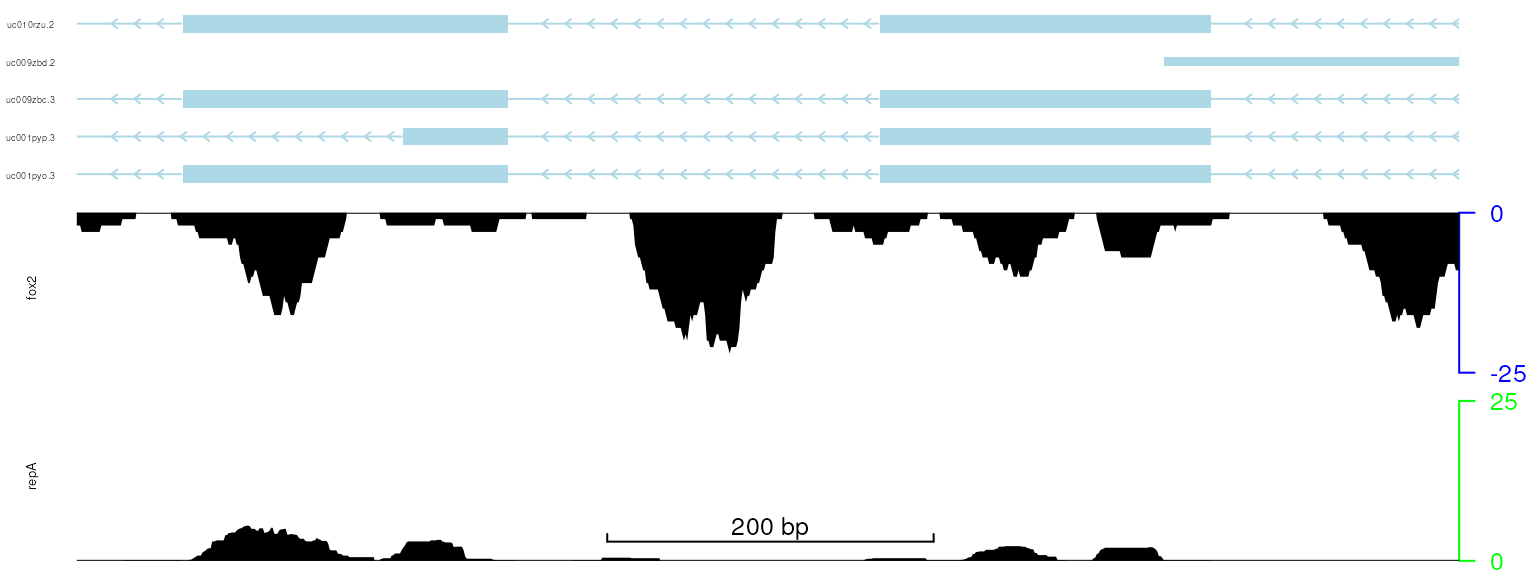
plot data with adjusted track color
Adjust the label of y-axis
The style of y-axis can be changed by setting the ylabgp slot in the style of each track.
setTrackStyleParam(trackList[[1]], "ylabgp", list(cex=.8, col="green"))
## set cex to avoid automatic adjust
setTrackStyleParam(trackList[[2]], "ylabgp", list(cex=.8, col="blue"))
setTrackStyleParam(trackList[[2]], "marginBottom", .2)
viewTracks(trackList, gr=gr, viewerStyle=viewerStyle)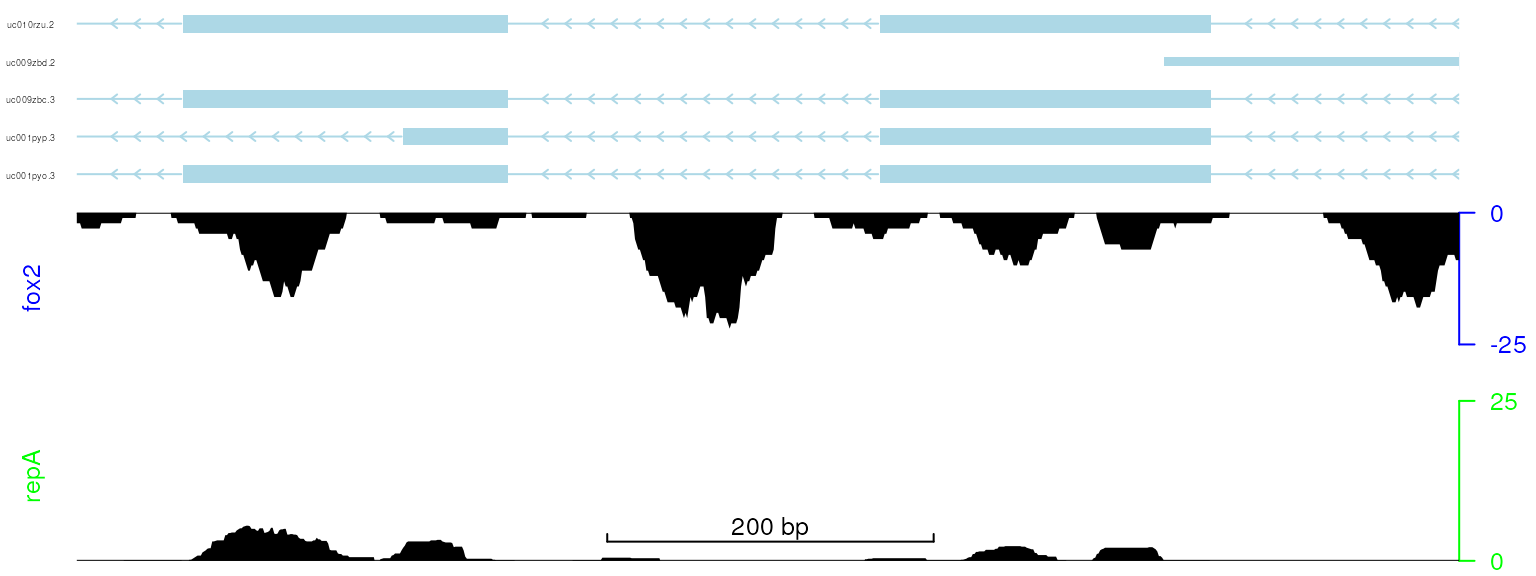
plot data with adjusted color and size of y-axis label
The y-axis label can be put at the top or the bottom of each track.
setTrackStyleParam(trackList[[1]], "ylabpos", "bottomleft")
setTrackStyleParam(trackList[[2]], "ylabpos", "topright")
setTrackStyleParam(trackList[[2]], "marginTop", .2)
viewTracks(trackList, gr=gr, viewerStyle=viewerStyle)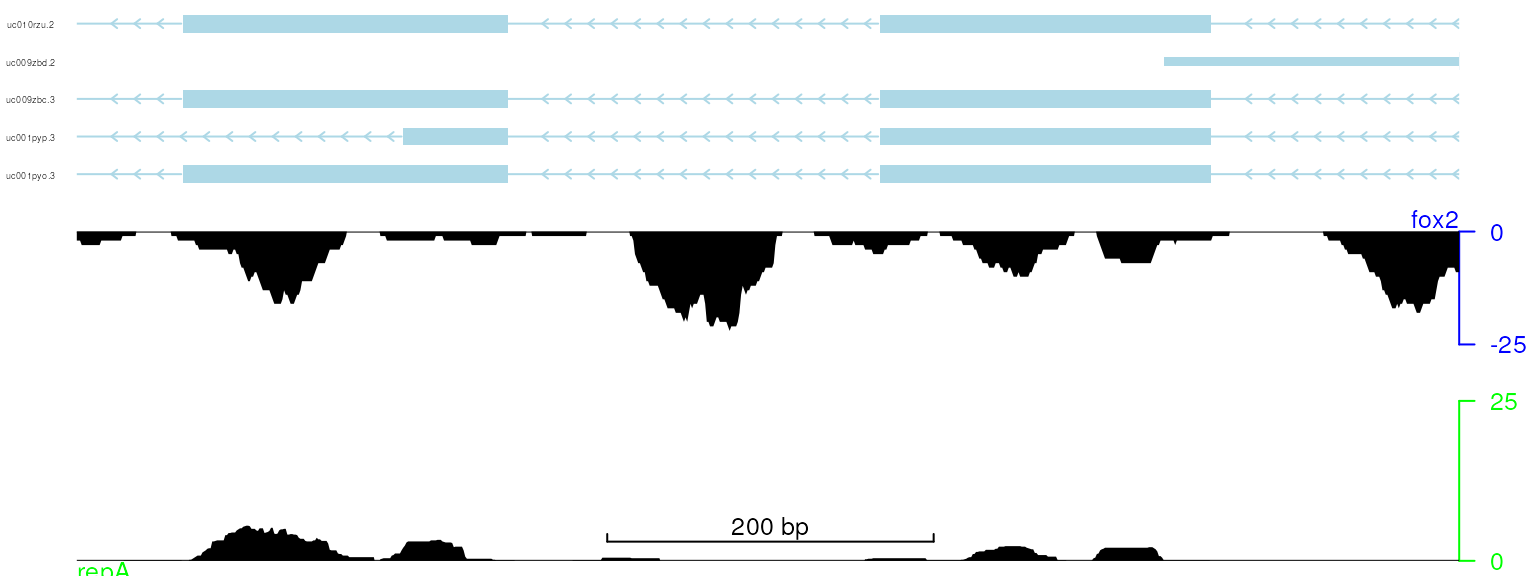
plot data with adjusted y-axis label position
For each transcript, the transcript name can be put on the upstream or downstream of the transcript.
trackListN <- trackList
setTrackStyleParam(trackListN[[3]], "ylabpos", "upstream")
setTrackStyleParam(trackListN[[4]], "ylabpos", "downstream")
## set cex to avoid automatic adjust
setTrackStyleParam(trackListN[[3]], "ylabgp", list(cex=.6))
setTrackStyleParam(trackListN[[4]], "ylabgp", list(cex=.6))
gr1 <- range(unname(unlist(GRangesList(sapply(trs, function(.ele) .ele$dat)))))
start(gr1) <- start(gr1) - 2000
end(gr1) <- end(gr1) + 2000
viewTracks(trackListN, gr=gr1, viewerStyle=viewerStyle)
plot data with adjusted transcripts name position
Adjust the track color
The track color can be changed by setting the color slot in the style of each track. The first color is for the dat slot of track and the second color is for the dat2 slot.
setTrackStyleParam(trackList[[1]], "color", c("green", "black"))
setTrackStyleParam(trackList[[2]], "color", c("black", "blue"))
for(i in 3:length(trackList))
setTrackStyleParam(trackList[[i]], "color", "black")
viewTracks(trackList, gr=gr, viewerStyle=viewerStyle)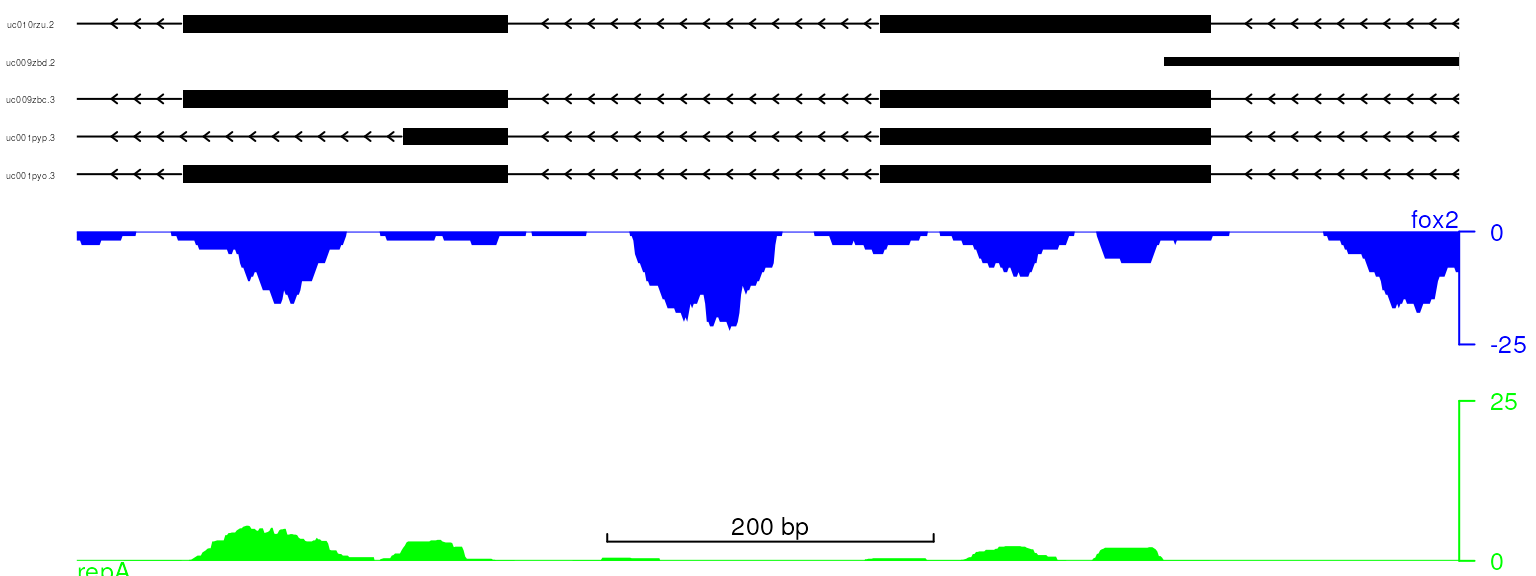
plot data with adjusted track color
Adjust the track height
The track height can be changed by setting the height slot in the style of each track. However, the total height for all the tracks should be 1.
trackListH <- trackList
setTrackStyleParam(trackListH[[1]], "height", .1)
setTrackStyleParam(trackListH[[2]], "height", .44)
for(i in 3:length(trackListH)){
setTrackStyleParam(trackListH[[i]], "height",
(1-(0.1+0.44))/(length(trackListH)-2))
}
viewTracks(trackListH, gr=gr, viewerStyle=viewerStyle)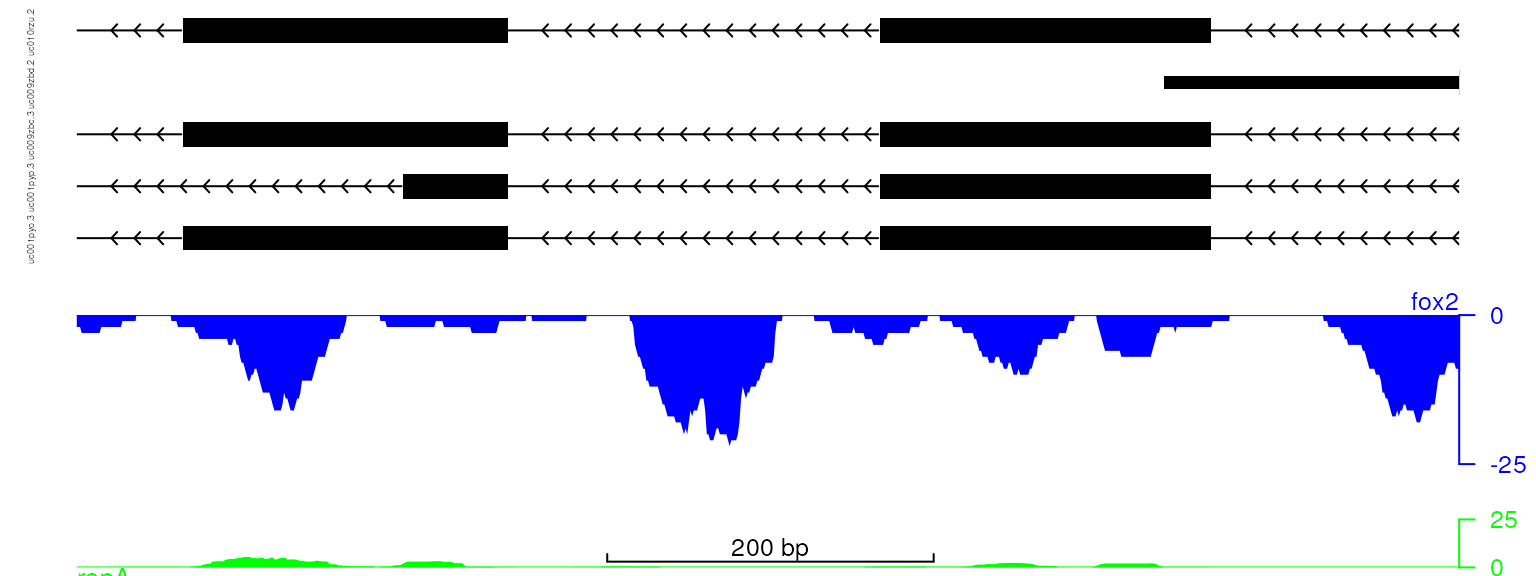
plot data with adjusted track height
Change the track names
The track names such as gene model names can be edited easily by changing the names of trackList.
names(trackList) <- c("cpsf160", "fox2", rep("HSPA8", 5))
viewTracks(trackList, gr=gr, viewerStyle=viewerStyle)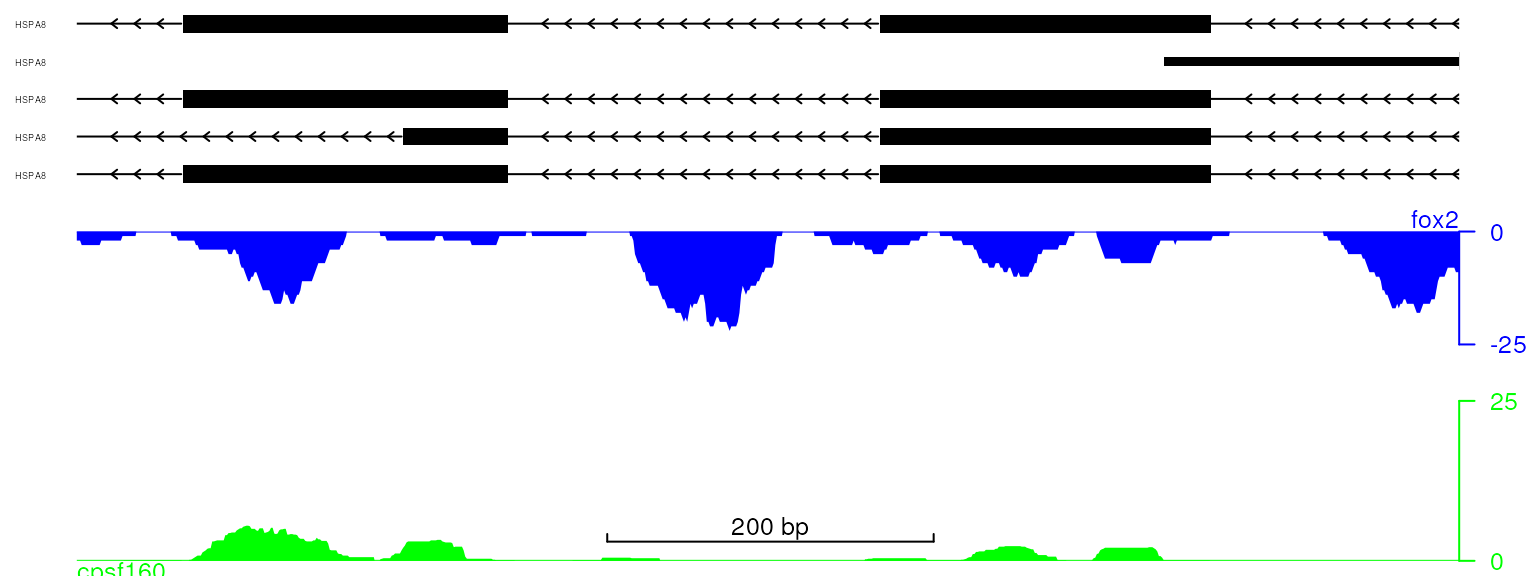
change the track names
Show paired data in the same track
trackViewer can be used to show to-be-compared data in the same track side by side.
cpsf160 <- importScore(file.path(extdata, "cpsf160.repA_-.wig"),
file.path(extdata, "cpsf160.repB_-.wig"),
format="WIG")
strand(cpsf160$dat) <- strand(cpsf160$dat2) <- "-"
setTrackStyleParam(cpsf160, "color", c("black", "red"))
viewTracks(trackList(trs, cpsf160), gr=gr, viewerStyle=viewerStyle)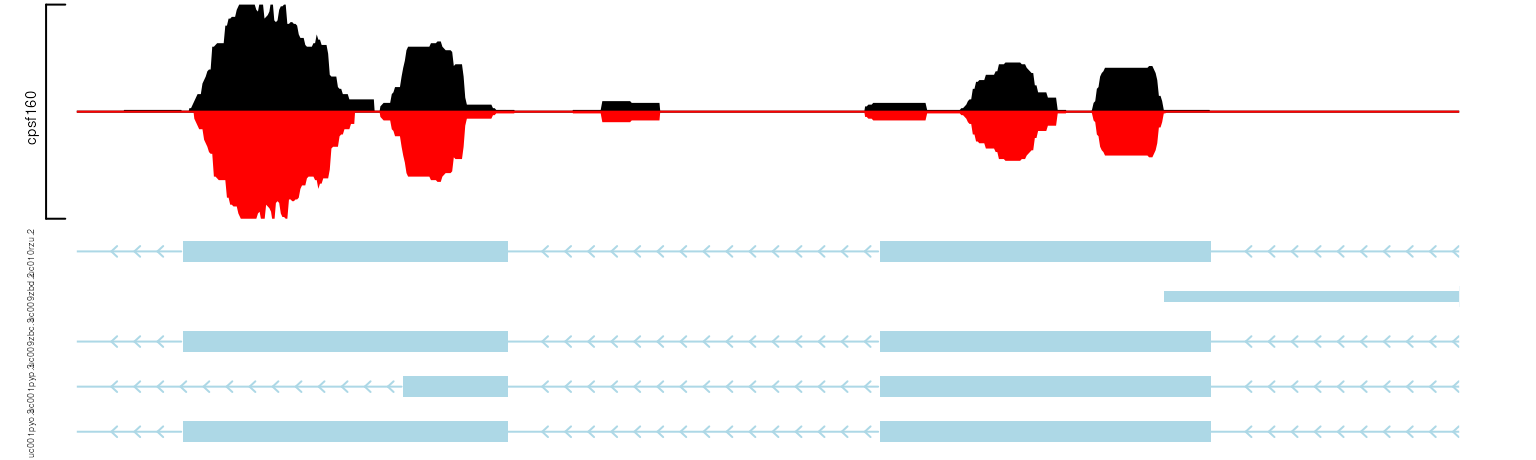
show two data in the same track
Flip the x-axis
The x-axis can be horizotally flipped for the genes in the negative strand.
viewerStyleF <- viewerStyle
setTrackViewerStyleParam(viewerStyleF, "flip", TRUE)
setTrackViewerStyleParam(viewerStyleF, "xaxis", TRUE)
setTrackViewerStyleParam(viewerStyleF, "margin", c(.1, .05, .01, .01))
vp <- viewTracks(trackList, gr=gr, viewerStyle=viewerStyleF)
addGuideLine(c(122929767, 122929969), vp=vp)
addArrowMark(list(x=122929650,
y=2),
label="label",
col="blue",
vp=vp)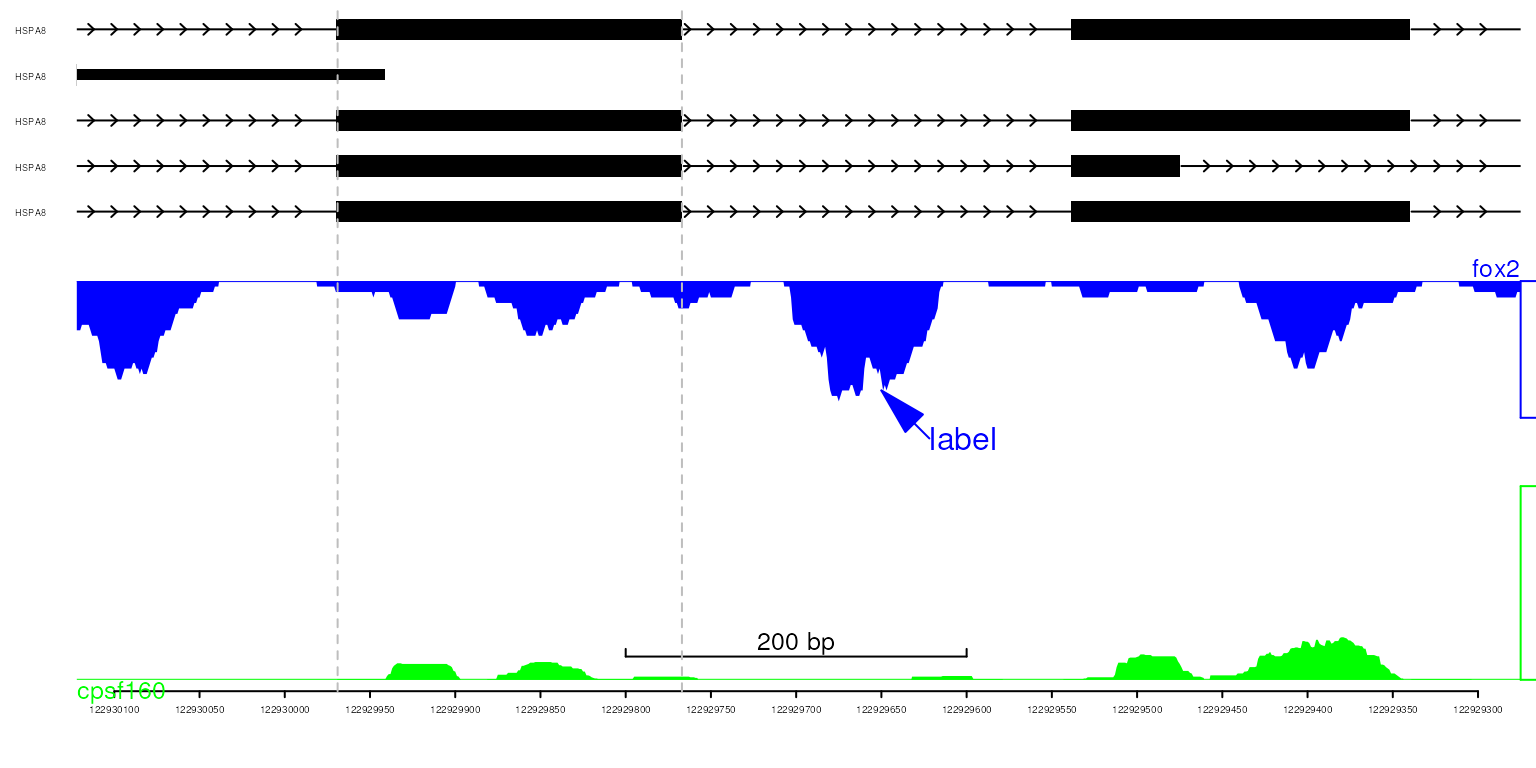
show data in the flipped track
Optimize the theme
Currently, we support two themes: bw (black and white) and col (colored).
optSty <- optimizeStyle(trackList(repA, fox2, trs), theme="bw")
trackList <- optSty$tracks
viewerStyle <- optSty$style
vp <- viewTracks(trackList, gr=gr, viewerStyle=viewerStyle)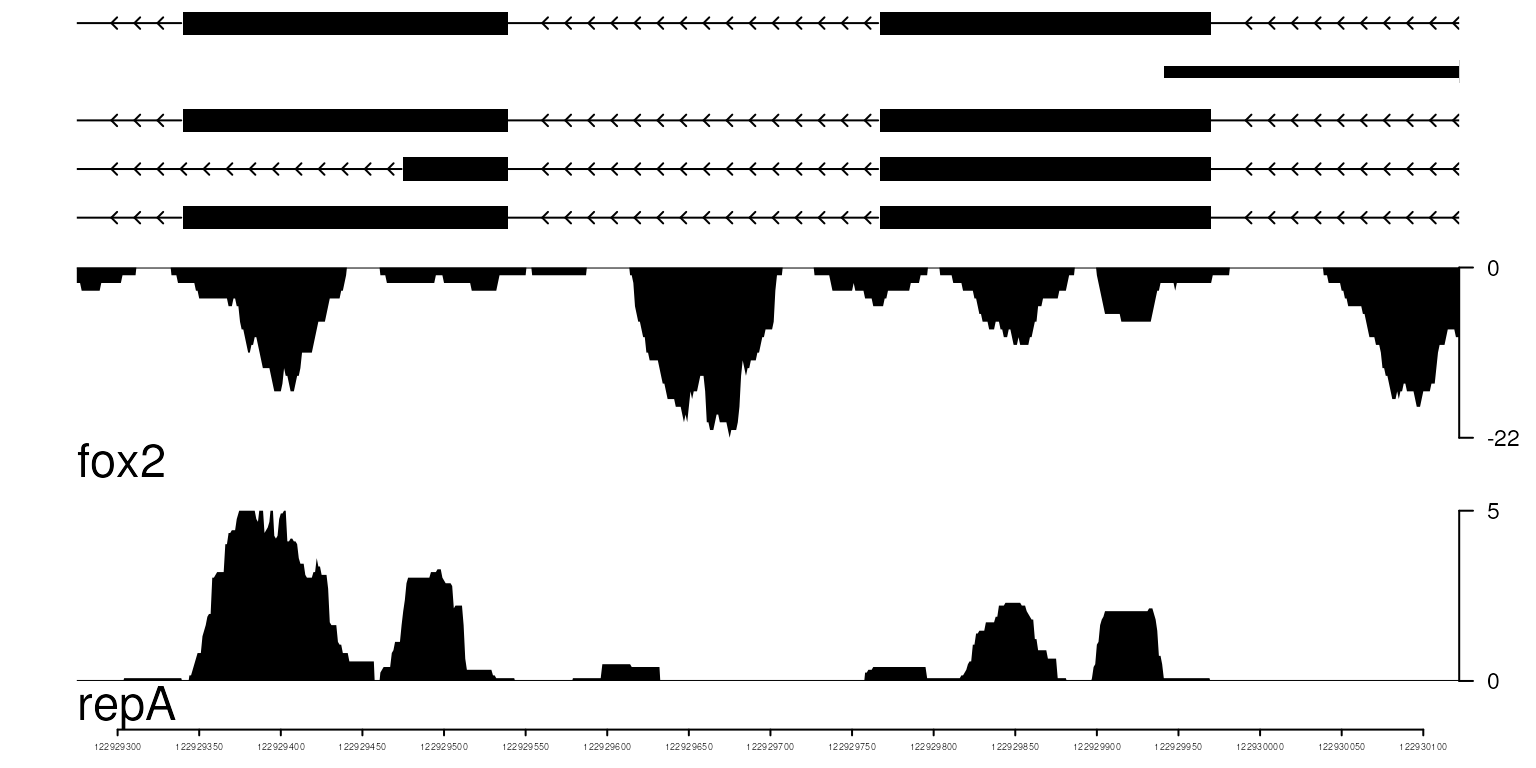
balck & white theme
optSty <- optimizeStyle(trackList(repA, fox2, trs), theme="col")
trackList <- optSty$tracks
viewerStyle <- optSty$style
vp <- viewTracks(trackList, gr=gr, viewerStyle=viewerStyle)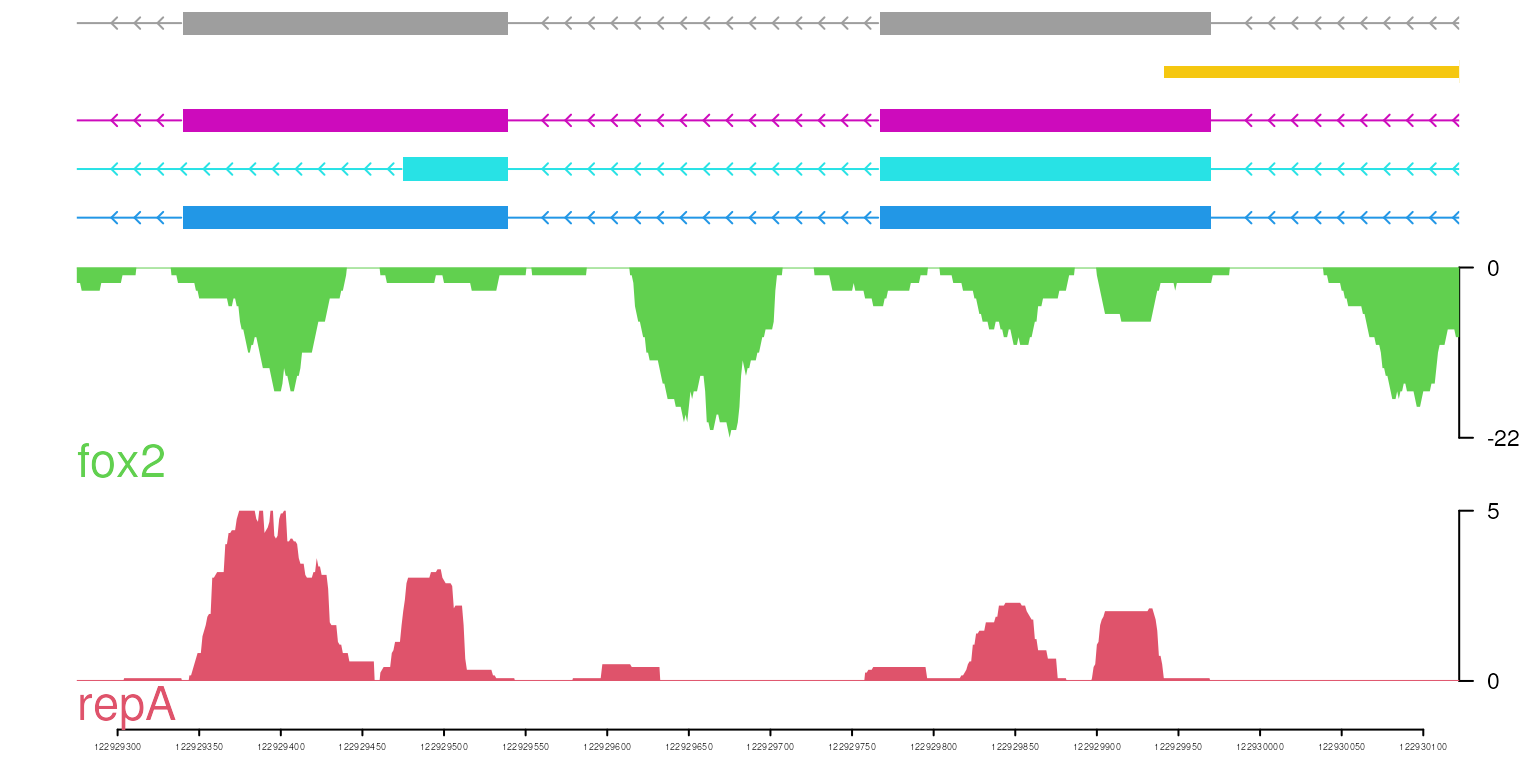
colorful theme
optSty <- optimizeStyle(trackList(repA, fox2, trs), theme="safe")
trackList <- optSty$tracks
viewerStyle <- optSty$style
vp <- viewTracks(trackList, gr=gr, viewerStyle=viewerStyle)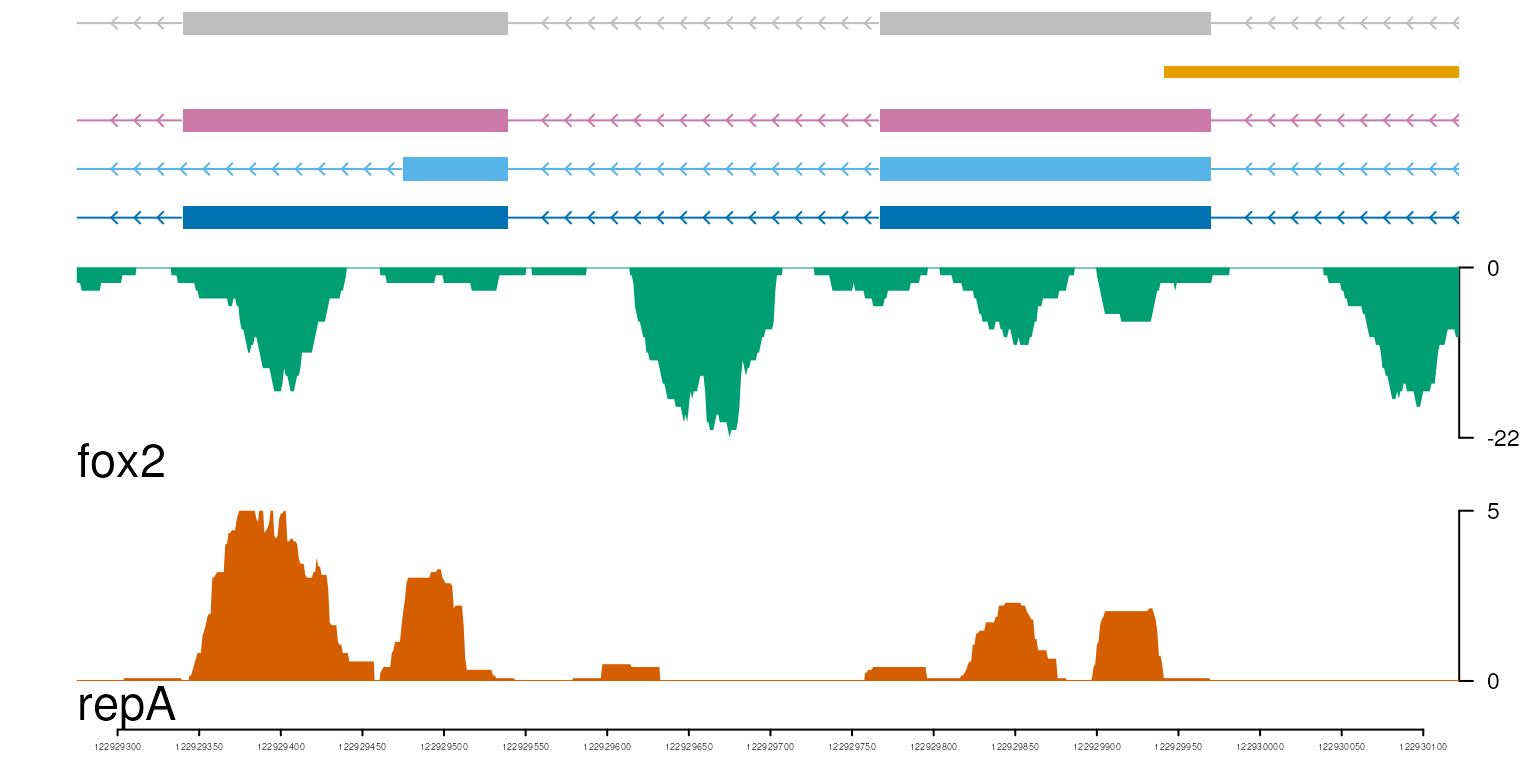
safe theme
Plot with breaks
We could plot the tracks with breaks by setting multiple genomic ranges.
gr.breaks <- GRanges("chr11",
IRanges(c(122929275, 122929575, 122929775),
c(122929555, 122929725, 122930122)),
strand="-", percentage=c(.4, .2, .4))
vp <- viewTracks(trackList, gr=gr.breaks, viewerStyle=viewerStyle)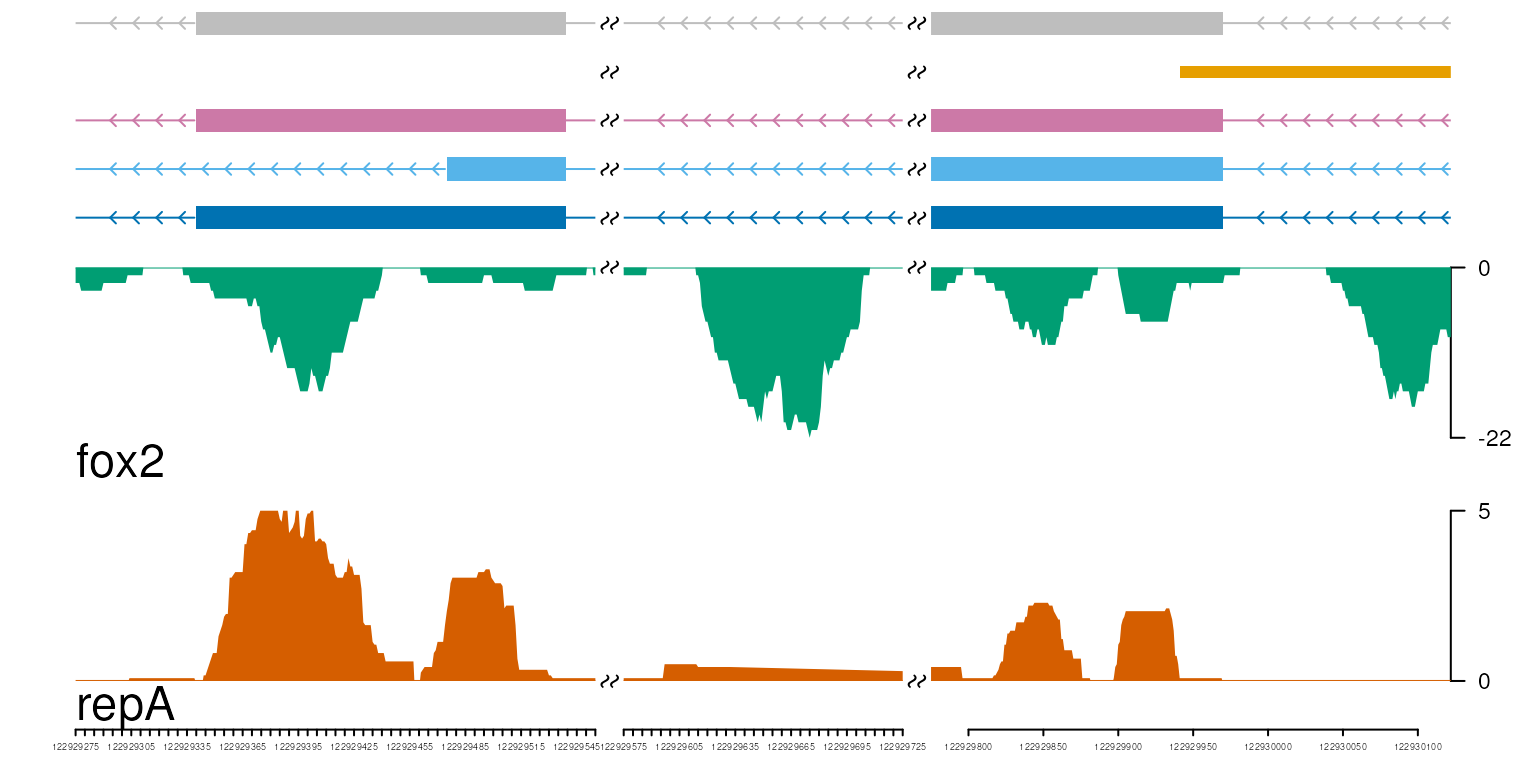
axis with breaks
Session Info
R version 4.4.1 (2024-06-14) Platform: x86_64-pc-linux-gnu Running under: Ubuntu 22.04.4 LTS
Matrix products: default BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3 LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.20.so; LAPACK version 3.10.0
locale: [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
time zone: Etc/UTC tzcode source: system (glibc)
attached base packages: [1] grid stats4 stats graphics grDevices utils datasets [8] methods base
other attached packages: [1] httr_1.4.7
[2] VariantAnnotation_1.51.0
[3] Rsamtools_2.21.1
[4] Biostrings_2.73.1
[5] XVector_0.45.0
[6] SummarizedExperiment_1.35.1
[7] MatrixGenerics_1.17.0
[8] matrixStats_1.4.0
[9] org.Hs.eg.db_3.19.1
[10] TxDb.Hsapiens.UCSC.hg19.knownGene_3.2.2 [11]
GenomicFeatures_1.57.0
[12] AnnotationDbi_1.67.0
[13] Biobase_2.65.1
[14] Gviz_1.49.0
[15] rtracklayer_1.65.0
[16] trackViewer_1.41.6
[17] GenomicRanges_1.57.1
[18] GenomeInfoDb_1.41.1
[19] IRanges_2.39.2
[20] S4Vectors_0.43.2
[21] BiocGenerics_0.51.1
loaded via a namespace (and not attached): [1] strawr_0.0.92
RColorBrewer_1.1-3 rstudioapi_0.16.0
[4] jsonlite_1.8.8 magrittr_2.0.3 rmarkdown_2.28
[7] fs_1.6.4 BiocIO_1.15.2 zlibbioc_1.51.1
[10] ragg_1.3.2 vctrs_0.6.5 memoise_2.0.1
[13] RCurl_1.98-1.16 base64enc_0.1-3 htmltools_0.5.8.1
[16] S4Arrays_1.5.7 progress_1.2.3 curl_5.2.2
[19] Rhdf5lib_1.27.0 rhdf5_2.49.0 SparseArray_1.5.31
[22] Formula_1.2-5 sass_0.4.9 bslib_0.8.0
[25] htmlwidgets_1.6.4 desc_1.4.3 httr2_1.0.3
[28] cachem_1.1.0 GenomicAlignments_1.41.0 lifecycle_1.0.4
[31] pkgconfig_2.0.3 Matrix_1.7-0 R6_2.5.1
[34] fastmap_1.2.0 GenomeInfoDbData_1.2.12 digest_0.6.37
[37] colorspace_2.1-1 textshaping_0.4.0 Hmisc_5.1-3
[40] RSQLite_2.3.7 filelock_1.0.3 fansi_1.0.6
[43] abind_1.4-5 compiler_4.4.1 bit64_4.0.5
[46] htmlTable_2.4.3 backports_1.5.0 BiocParallel_1.39.0
[49] DBI_1.2.3 highr_0.11 biomaRt_2.61.3
[52] rappdirs_0.3.3 DelayedArray_0.31.11 rjson_0.2.22
[55] tools_4.4.1 foreign_0.8-87 nnet_7.3-19
[58] glue_1.7.0 InteractionSet_1.33.0 restfulr_0.0.15
[61] rhdf5filters_1.17.0 checkmate_2.3.2 cluster_2.1.6
[64] generics_0.1.3 gtable_0.3.5 BSgenome_1.73.0
[67] ensembldb_2.29.1 data.table_1.16.0 hms_1.1.3
[70] xml2_1.3.6 utf8_1.2.4 pillar_1.9.0
[73] stringr_1.5.1 dplyr_1.1.4 BiocFileCache_2.13.0
[76] lattice_0.22-6 deldir_2.0-4 bit_4.0.5
[79] biovizBase_1.53.0 tidyselect_1.2.1 knitr_1.48
[82] gridExtra_2.3 ProtGenerics_1.37.1 xfun_0.47
[85] stringi_1.8.4 UCSC.utils_1.1.0 lazyeval_0.2.2
[88] yaml_2.3.10 evaluate_0.24.0 codetools_0.2-20
[91] interp_1.1-6 tibble_3.2.1 BiocManager_1.30.25
[94] cli_3.6.3 rpart_4.1.23 systemfonts_1.1.0
[97] munsell_0.5.1 jquerylib_0.1.4 Rcpp_1.0.13
[100] dichromat_2.0-0.1 grImport_0.9-7 dbplyr_2.5.0
[103] png_0.1-8 XML_3.99-0.17 parallel_4.4.1
[106] pkgdown_2.1.0 ggplot2_3.5.1 blob_1.2.4
[109] prettyunits_1.2.0 latticeExtra_0.6-30 jpeg_0.1-10
[112] AnnotationFilter_1.29.0 bitops_1.0-8 txdbmaker_1.1.1
[115] scales_1.3.0 crayon_1.5.3 BiocStyle_2.33.1
[118] rlang_1.1.4 KEGGREST_1.45.1